快速发展的测序技术产生了大量背景复杂、内涵丰富的数据,如何从纷繁复杂的生物信息数据中恢复出数据的真相,打开认知生物奥秘的窗口,成为当今人工智能与相关交叉学科研究的前沿课题。其中,单细胞蛋白组数据独特的复杂性使得蛋白组数据的分析处理成为一个严峻的挑战。单细胞蛋白组中存在着独特的批次效应(内部批次效应与外部批次效应)、数据噪声和数据缺失等问题。这些数据问题在蛋白组数据的处理过程中互相交织、相互影响,而针对转录组数据的分析方法并不能完全解决这些问题。随着单细胞蛋白组技术的蓬勃发展,迫切需要解决这一系列数据问题的完整方法。
3月19日,南开大学人工智能学院张瀚团队联合腾讯AI实验室姚建华团队在国际顶级学术期刊《自然•方法》(Nature Methods)上发表题为“scPROTEIN: 一种用于单细胞蛋白质组学嵌入的多功能深度图对比学习框架”(scPROTEIN: a versatile deep graph contrastive learning framework for single-cell proteomics embedding) 的文章。该研究提出一种名为scPROTEIN的基于图对比学习的单细胞蛋白质组学表征学习方法,此方法首次开发了一个统一的深度学习框架,以解决质谱测序带来的数据缺失、批次效应和高噪声等在数据处理中互相影响的难题,并学习到准确的细胞嵌入表示,可用于一系列下游分析。
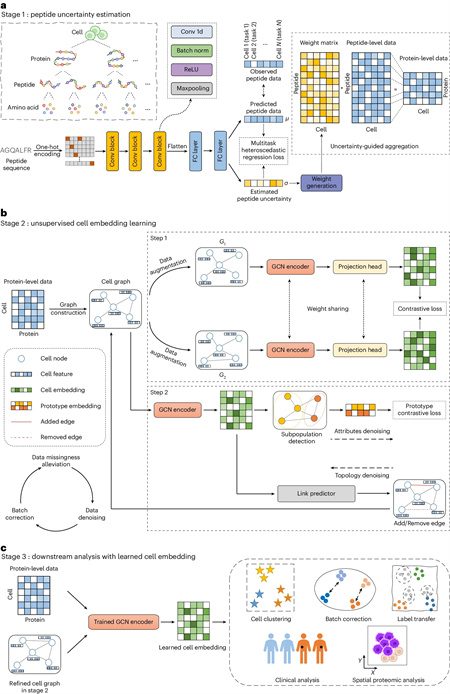
scPROTEIN模型示意图
scPROTEIN是针对于单细胞蛋白质组学数据的计算方法。模型首先通过多任务异方差回归模型为每个细胞中的每个肽信号分配不确定性权重,并设计不确定性引导的方式从肽水平丰度聚合得到蛋白质水平丰度,充分利用了蛋白质组学的分层结构,提高了数据质量。scPROTEIN通过建立细胞图结构来表征单细胞蛋白质组学数据,在图上的消息传递过程充分考虑了共表达模式以缓解数据缺失问题。模型基于图对比学习进行自监督训练,并且对比学习的判别性可以在不依赖数据先验知识的情况下隐式地去除批次效应。scPROTEIN 同时设计了属性-拓扑迭代去噪模块,可以对蛋白质组数据进行去噪并得到准确的细胞表征。属性降噪基于原型对比学习,利用远离边界的细胞原型来增强其他细胞的信息。增强后的细胞表征用于动态完善细胞图拓扑结构,并有助于进一步得到更准确的细胞表征。
通过一系列下游任务上的综合实验,文章系统地展示了scPROTEIN在基于质谱和基于抗体测序的蛋白质组学上的适用性和优异性能。与现有的单细胞蛋白质组学数据处理流程和其他转录组对比方法相比,scPROTEIN在细胞聚类、批次校正和细胞类型注释任务中表现出更优越的性能。此外,scPROTEIN在单细胞临床蛋白组分析和空间分辨率的蛋白质数据分析中均展现出广泛的适用性。随着单细胞蛋白质组学技术的迅速发展和应用,scPROTEIN 将在各类单细胞蛋白质组学数据分析场景中发挥越来越重要的作用,为解读复杂的生物数据提供了新的方法与工具。这一前沿成果为科学智能(AI for Science)提供了新的思路,显示了深度学习在解决生物医学数据分析中复杂问题的巨大潜力。
南开大学为论文第一完成单位,南开大学博士生李威为第一作者,南开大学人工智能学院张瀚教授和腾讯AI实验室姚建华博士为共同通讯作者,腾讯AI实验室杨帆博士为并列第一作者。相关研究得到了国家自然科学基金资助。
文章链接:https://www.nature.com/articles/s41592-024-02214-9

